Bitte bestätigen Sie, dass Sie mit den Inhalten unserer Datenschutzerklärung einverstanden sind sowie dem Einsatz von Cookies zustimmen. Im Bereich "Cookie-Managment" in unserer Datenschutzerklärung können Sie Cookies jederzeit aktivieren oder deaktivieren.

Fakten zum Thema LAM
Bitte ein Thema auswählen...:
Was ist LAM?
Subtitle Title or Description
Lymphangioleiomyomatose oder LAM ist eine sehr seltene Erkrankung, die fast ausschließlich Frauen betrifft und häufig im Alter zwischen 30 und 45 Jahren festgestellt wird. In der medizinischen Literatur wurde LAM erstmals von Stossel 1937 beschrieben.
LAM betrifft hauptsächlich die Lungen. Dort verursacht sie ein übermäßiges Wachstum der so genannten glatten Muskelzellen. Obwohl diese Zellen unter dem Mikroskop nicht wie Krebszellen erscheinen, gibt es einige Ähnlichkeiten zu Krebs, im Wesentlichen:
- Die LAM Zellen wachsen unkontrolliert
- Es gibt die Hypothese, dass die LAM Zellen in die Lunge metastieren, wobei der Ursprung noch unklar ist.
Ein wesentlicher Unterschied zu Krebs ist vor allem die Tatsache, dass LAM gewöhnlicherweise langsam, über oft viele Jahre hinweg progredient und weniger aggressiv verläuft.
Weitere Informationen dazu: http://www.thelamfoundation.org/LAM-Patients-Family-Friends/Resources/LAM-as-Cancer
Das vermehrte Zellwachstum tritt entlang der Luftwege auf, sowie entlang der Blutgefäße, in den Wänden der Lymphgefäße und in bestimmten Teilen des Lungengewebes. Im Laufe der Zeit bilden diese Zellen Ansammlungen und wachsen in die Wände der Luftwege und Lymphbahnen, und bilden dort Hindernisse.
Die LAM verändert das Lungengewebe, so dass zystenartige Strukturen das gesunde Lungengewebe immer mehr zerstören. Dadurch wird die Atmung stark beeinträchtigt und der Körper kann nicht mehr ausreichend mit Sauerstoff versorgt werden.
LAM wirkt sich hauptsächlich auf die Lunge aus, jedoch haben bis zu 50% der an LAM erkrankten Frauen auch Angiomyolipome. Diese Nierentumore sind nicht bösartig, können aber Blutungen verursachen. Bei Patienten mit LAM können auch häufiger fibrotische Gewebeveränderungen im Bauchraum und vergrößerte Lymphknoten auftreten.
Der Name Lymphangioleiomyomatose verdeutlicht die verschiedenen Komponenten der Krankheit. Lymph und angio beziehen sich auf die Lymph- und Blutgefäße, die betroffen sind, und leiomyomatose bezieht sich auf die glatten Muskelzellen.
Es ist allerdings eine genetische Verbindung zu einer anderen Krankheit, der Tuberösen Sklerose entdeckt worden. Daher gehen Forscher jetzt eher von 250.000 an LAM erkrankten Frauen weltweit aus. (Quelle: LAM Foundation, USA)
Es gibt zwei Typen von LAM: die so genannte sporadische LAM (engl.: sporadic LAM), die nicht vererbt werden kann, und LAM, die im Zusammenhang mit der Erkrankung Tuberöser Sklerose auftritt und vererbbar ist.
Was sind die Symptome von LAM?
Subtitle Title or Description
Es gibt verschiedene Anzeichen für LAM.
Gewöhnlich bemerken die Patientinnen zuerst eine Kurzatmigkeit bei Belastung. Dies kann im frühen Stadium von LAM erstmals bei starker Anstrengung, z.B. beim Sport auffallen. Mit dem Fortschreiten der Krankheit tritt die Kurzatmigkeit dann auch bei alltäglicher Belastung auf, beispielsweise beim Treppensteigen. Die Einschränkung der Atmung wird durch die Veränderungen im Lungengewebe und in den Luftwegen verursacht.
Weitere häufige Symptome sind Schmerzen im Brustkorb und Husten. Da LAM auch die Blutbahnen betreffen kann, wird von manchen Patienten auch gelegentlich Blut gehustet.
Für viele Patienten mit LAM ist das erste deutliche Anzeichen der Krankheit ein Lungenkollaps (Spontan-Pneumothorax). Dieser tritt dann auf, wenn Gewebe an der Lungenoberfläche aufbricht und Luft aus der Lunge in den Brustkorb gelangt. Bei einem Pneumothorax tritt die Kurzatmigkeit meistens plötzlich ein und ist oft von stechenden Schmerzen begleitet. Der Pneumothorax kann mittels einer Röntgenaufnahme des Brustkorbs festgestellt werden und erfordert eine Behandlung im Krankenhaus. Tritt der Pneumothorax wiederholt auf, kann eine Operation notwendig werden, um weitere Pneumothoraces zu verhindern. Neuere Empfehlungen geben den Hinweis, bei einer diagnostizierten LAM bereits nach dem 1. Pneumothorax mit einer Pleurodese zu behandeln [(s. Leitlinien 2017). ]((s. Leitlinien 2017)
Ein häufiges Symptom ist auch der Chylothorax, bei dem sich Lymphflüssigkeit um die Lunge herum ansammelt (pleuraler Erguß) und die Lunge einengt. Dies tritt dann auf, wenn durch die Vermehrung der LAM-Zellen die Lymphbahnen blockiert werden. Wenn sich sehr viel Lymphflüssigkeit im Brustkorb ansammelt, ist eine chirurgische Behandlung notwendig, um die Flüssigkeit zu entfernen.
Wie wird LAM festgestellt?
Subtitle Title or Description
Viele Symptome von LAM ähneln denen von Asthma, chronischer Bronchitis oder von einem Lungenemphysem. Deshalb kommt es vor, dass Patienten bereits lange Zeit Beschwerden haben, bevor LAM festgestellt wird. Die durchschnittliche Zeit von den ersten Symptomen und der Diagnose beträgt 3–6 Jahre. Die Symptome, Röntgenbilder und die Lungenfunktion können bereits auf LAM deuten, aber die sichere Diagnose wird meistens durch eine HRCT (hochauflösende Computertomografie) der Lungen und/oder mittels einer Lungenbiopsie (Gewebeentnahme) gestellt. Bei der computertomografischen Aufnahme (HRCT) der Lunge sind die für LAM typischen blasenartigen Veränderungen des Lungengewebes deutlich zu erkennen. Manchmal ist auch eine Lungenbiopsie erforderlich. Dabei wird Gewebe aus der Lunge entnommen und auf bestimmte Zellveränderungen untersucht. Auf diese Weise lässt sich LAM eindeutig diagnostizieren.
Hier der Diagnostische Algorithmus (aus: https://en.wikipedia.org/wiki/Lymphangioleiomyomatosis)
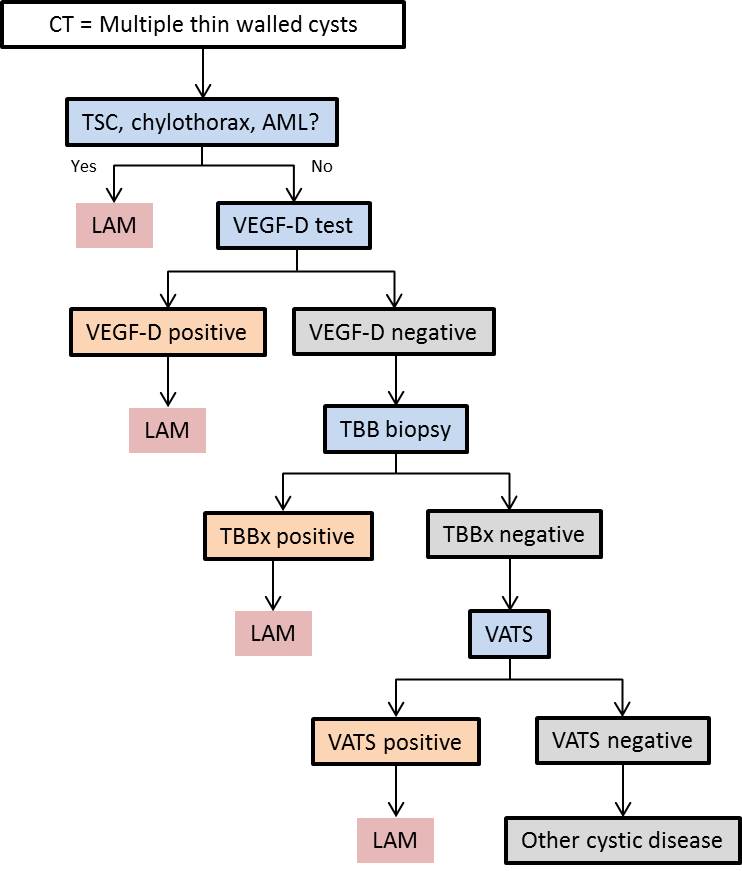
NEU! Die Messung des VEGF (Vascular Endothelial Growth Factor-D) zur Unterstützung der Erstdiagnose und Therapiekontrolle ist am Universitätsklinikum Leipzig ab sofort verfügbar!
Hier die Information zur Einsendung von Proben: https://www.lam-info.de/cms-data/depot/lam-depot/Hinweise%5FVEGF%5FD%5F%5Fexterne%5FEinsender%5FStand%5F20170815.pdf
Wie verläuft LAM?
Subtitle Title or Description
LAM ist eine progressive Erkrankung, d.h. es ist mit einer allmählichen Verschlechterung der Lungenfunktion zu rechnen, wobei die Verläufe unterschiedlich sein können. Typischerweise entwickeln LAM-Patientinnen eine sich stetig verschlechternde Obstruktion der Atemwege. Allerdings ist der Verlauf in der Regel langsam und die Prognose hat sich in den letzten Jahren stetig verbessert. In einer neueren Studie wurde ein mittleres Überleben mit 29Jahren angegeben, was eine deutlich optimistischere Prognose im Vergleich zu früheren Studien darstellt. Hier die Grafik aus der Präsentation von Prof. Wirtz einfügen.
Mit dem Fortschreiten der Erkrankung vermehrt sich das Wachstum der glatten Muskelzellen in der Lunge und das ursprüngliche Lungengewebe wird immer mehr durch blasenartige Strukturen ersetzt. Das Lungengewebe nimmt ein wabenartiges Aussehen an. Im schlimmsten Fall kann davon die ganze Lunge betroffen sein. Ein weiteres Problem ist, dass LAM-Zellschichten in den Lungenbläschen und Bronchien den Luftstrom behindern (Obstruktion). Außerdem wird der Sauerstoffaustausch durch die Zellwände hindurch erschwert, weil sich zusätzliche Zellschichten auf der dünnen Wand der Lungenbläschen bilden. Durch diese Veränderungen wird die Atmung immer mehr beeinträchtigt. Deshalb wird für viele Patienten im Verlauf der Erkrankung eine Sauerstofftherapie nötig.
Wie wird LAM behandelt?
Subtitle Title or Description
Medication
Sirolimus is an mTOR inhibitor that stabilizes lung function and improves some measures of life in LAM patients.[35] It is approved by the FDA for use in LAM, based on the results of the Multicenter International LAM Efficacy and Safety of Sirolimus (MILES) Trial. MILES data supports the use of sirolimus in patients who have abnormal lung function (i.e. FEV1<70% predicted). Whether the benefits of treatment outweigh the risks for asymptomatic LAM patients with normal lung function is not clear, but some physicians consider treatment for declining patients who are approaching the abnormal range for FEV1. Sirolimus also appears to be effective for the treatment chylous effusions and lymphangioleiomyomatosis. The benefits of sirolimus only persist while treatment continues. The safety of long term therapy has not been studied.
Potential side effects from mTOR inhibitors include swelling in the ankles, acne, oral ulcers, dyspepsia, diarrhea, elevation of cholesterol and triglycerides, hypertension and headache. Sirolimus pneumonitis and latent malignancy are more serious concerns, but occur infrequently. Sirolimus inhibits wound healing. It is important to stop therapy with the drug for 1–2 weeks before and after elective procedures that require optimal wound healing. Precautions must be taken to avoid prolonged sun exposure due to increased skin cancer risk.
Treatment with another mTOR inhibitor, everolimus, was reported in a small, open-label trial to be associated with improvement in FEV1 and six-minute walk distance.[125] Serum levels of VEGF-D and collagen IV were reduced by treatment. Adverse events were generally consistent with those known to be associated with mTOR inhibitors, although some were serious and included peripheral edema, pneumonia, cardiac failure and Pneumocystis jirovecii infection. Escalating doses of everolimus were used, up to 10 mg per day; higher than what is typically used clinically for LAM.
Serum VEGF-D concentration is useful, predictive and prognostic biomarker.[71] Higher baseline VEGF-D levels predicts more rapid disease progression and a more robust treatment response.
Hormonal approaches to treatment have never been tested in proper trials. In the absence of proven benefit, therapy with progesterone, GnRh agonists (e.g., Lupron, goserelin) and tamoxifen are not routinely recommended. Doxycycline had no effect on the rate of lung function decline in a double blind trial.[126].
Sirolimus is often effective as first-line management for chylothorax.[118] If chylous leakage or accumulations persist despite treatment, imaging with heavy T2 weighted MRI, MRI lymphangiography or thoracic duct lymphangiography can be considered. Pleural fusion procedures can be considered in refractory cases.[citation needed]
LAM Management
Subtitle Title or Description
There are several options in managing LAM. Patients and physicians should make therapy decisions jointly after thorough discussion of the risks and benefits of all options.
mTOR Inhibitor Therapy
Long-term treatment with an mTOR inhibitor is generally recommended for patients who have abnormal lung function and evidence of progressive lung function decline. mTOR therapy is also effective in patients with refractory chylous effusions. Trials are planned to determine if early low dose sirolimus therapy can prevent progression to more advanced stages:
- RAPAMUNE® (sirolimus) is the first drug to be approved by the FDA for the treatment of LAM. It is a highly specific inhibitor of the mechanistic target of the rapamycin (mTOR) signaling pathway that is dysregulated in LAM cells. Treatment with the drug suppresses growth of LAM cells and likely shrinks them, without killing them. It also reduces bad behaviors of LAM cells, such as production of VEGF-D that likely enhances tissue damage and the spread of LAM through the body. Sirolimus has been shown to stabilize lung function and improve quality of life in patients with moderate to severe LAM.
- Afinitor (everolimus) is also used in the treatment of LAM, although it is not FDA approved for that purpose. Everolimus has been shown to be effective for the treatment of angiomyolipomas and subependymal giant cell astrocytomas in patients with tuberous sclerosis. It is a derivative of sirolimus and has very similar functions and side effects.
Supportive Measures
- Oxygen therapy may become necessary as LAM progresses and lung function becomes impaired. In addition, oxygen may prolong life in hypoxic patients. Oxygen should be administered to maintain arterial oxygen saturations of 90% or greater. It will also support rest, exercise and sleep and may help stabilize pulmonary hypertension.
- Bronchodilators may benefit patients. About 25% of LAM patients have an asthma-like component that responds to bronchodilators. Many LAM patients benefit from the use of drugs such as albuterol, formoterol and salmeterol.
Unproven Therapies
- Anti-estrogen therapies. Although patients with LAM have been managed empirically with anti-estrogen therapies for decades, there is no proof that they are effective. In addition, induction of early menopause is distressing and morbid in young women. Clinicians sometimes consider hormonal therapies in special situations, such as progression of lung function decline despite sirolimus use and in patients who have recurrent pneumothoraces that are timed with the menstrual cycle.
- Progestins can cause fluid retention and mood swings. They are no longer routinely used in patients with LAM.
- Gonadotrophin-releasing hormone (GnRH) antagonists (receptor blockers), such as LUPRON® (leuprolide), induce menopause. Their role in the treatment of LAM is unclear. The risk of bone and cardiac disease is increased.
- Corticosteroids, immunomodulatory cytotoxic agents or ovarian irradiation are not recommended.
- Oophorectomy (removal of the ovaries) is no longer routinely recommended in LAM patients. The procedure is invasive without clear evidence of efficacy. In addition, the risk of bone and heart disease is increased.
Lung Transplantation
Referral for evaluation for lung transplantation should be considered when the FEV1 approaches 30%. Patients may also be eligible based on other factors that profoundly affect quality of life, such as disabling shortness of breath or problems maintaining oxygen saturation despite high supplemental oxygen delivery rates. Both single and bilateral lung transplantation have been performed for patients with LAM and both are suitable options. One, five, and ten-year survival rates after lung transplantation are 89%, 67%, and 47%, respectively.
This content was created for general informational purposes only. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
© 2022 LAM Selbsthilfe Deutschland e.V.